
Abstract

INTRODUCTION
CE MARKING
The definition of an IVD MD is given in the Directive as:
‘In-vitro diagnostic medical device’ means any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, equipment, or system, whether used alone or in combination, intended by the manufacturer to be used in-vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information:
concerning a physiologicalor pathological state, or
concerning a congenital abnormality, or
to determine the safety and compatibility with potential recipients, or
to monitor therapeutic measures.
This definition implies that all instruments, reagents, calibrators, etc., used in professional laboratory medicine must be CE marked. Also specimen receptacles and manufactured devices that are used for medical analyses in a professional and commercial context, without being marketed, are subject to the Directive. However, products for general laboratory use, IVD MD's that are used for research purposes without any medical objectives, home made reagents which are not used commercially, and international certified reference materials and materials used for external quality assessment schemes, are not covered.
The total IVD MD market in Europe was about 5.6 billion US$ in 1998 and showed over the past four years a growth of about 4% per year (data from EDMA at www.edma-ivd.be). Most of the IVD market (87%) is concerned with IVD reagents, calibrators, etc., while the rest (13%) is concerned with IVD instruments. Approximately 70% of the IVD products on the European market are imported from the USA and Japan.
The global market of IVD products had a turnover of some 19 billion US$ in 1998.
In order to apply the CE mark, the manufacturer must declare that their product complies with all the “essential requirements” of the Directive. The essential requirements (Table II), are described in 43 paragraphs and compliance with all of them is mandatory.
ESSENTIAL REQUIREMENTS

Selected aspects of the general requirements need further explanation in order that a uniform implementation is achieved. The European Commission has mandated to the European Committee for Standardization (CEN), and the European Committee for Electro-technical Standardization (CENELEC), the development of some 22 standards, which include, for example, labeling and symbols, stability testing, colour codes for blood-specimen containers, traceability (see below), and “particular requirements for automatic and semiautomatic operating analytical equipment.” The standards are all under development and publication is expected in 2000 and later. When the European Commission judges them of significant importance, they will be published in the Official Journal of the European Communities, and from then on they will be called “harmonized” standards. Compliance with the harmonized standards is not compulsory and manufacturers may chose their own methods. However, in such cases the manufacturer must demonstrate that their methods are equivalent or better.
Compliance with the essential requirements of the IVD directive and the harmonized standards ensures end-users that the safety, reliability and quality of the IVD product is guaranteed. The application of the CE mark is for most IVD products the responsibility of the manufacturer (self-declaration), and must be underpinned by a valid “EC Declaration of Conformity.” However, before placing a product on the market, the manufacturer (or its authorized representative if the manufacturer does not have a registered place of business in the EC), shall notify the National Competent Authority in the country where it has its registered place of business. The notifications will be registered in a databank, which is accessible to the Competent Authorities in all Member States.
NOTIFIED BODIES
When the first draft of the IVD Directive was published in April 1995, discussions started about more strict requirements for IVD devices for HIV detection. Especially the events in France fueled this discussion and in the final version of the Directive, the IVD medical devices are divided in two groups:
IVDs for which the correct performance is essential to medical practice and the failure of which can cause a serious risk to health of patients
IVDs that do not constitute a direct risk to patients and are used by competently trained professionals, and for which the results obtained can often be confirmed by other means
The IVD MD's of the first category, where serious risk to health to patients is involved, have been subdivided in two further subclasses as described in Annex II of the Directive:
ANNEX II IVD MEDICAL DEVICES
List A products.
Reagents and reactive products, including related calibration and control materials for:
the determination of blood types ABO system, rhesus (C, c, D, E, e), anti-Kell
the detection, confirmation and quantification in human specimens of markers of HIV infection (HIV 1 and 2), HTLV I and II, and hepatitis B, C, and D
List B products.
Reagent and reagent products, including related calibrators and control materials, for:
determining the blood groups: anti-Duffy, anti-Kidd
determining irregular anti-erythrocytic antibodies
the detection and quantification in human samples of the congenital infections rubella and toxoplasmosis
diagnosing the hereditary disease phenylketonuria
determining the human infections cytomegalovirus and chlamydia
determining the HLA tissue groups DR, A, B
determining the tumor marker prostate specific antigen
the evaluation of the risk of trisomy 21 (including software)
the self diagnosis devices for the measurement of blood sugar
List A products and possibly also List B products, must comply with the Common Technical Specifications which have been drawn up by a team of experts and which are a further specification of the Essential Requirements as applied to these special products. A Common Technical Specification is not mandatory and a producer is allowed to use other specifications, provided that it can be demonstrated that they are equivalent or better than the Common Technical Specifications.
The IVDs that do not constitute a direct risk to patients have been subdivided into subclasses, which require different conformity assessment procedures:
IVDs for self-testing except those listed in Annex II
IVDs for performance evaluation
All other IVDs
A Notified Body plays an essential role in the conformity assessment procedure in all the above-mentioned categories, except for the groups “IVDs for performance evaluation” and “all other IVDs.” The role of the Notified Body may include approval of design dossiers and registration files, certification of the manufacturers ISO 9001 or 9002 quality system, and batch-release controls or EC verification. A Notified Body must be appointed by a National Competent Authority and must operate under their direct supervision. Any public or private organization that possesses the necessary technical, scientific and quality systems expertise can apply for nomination as a Notified Body. The different conformity assessment routes are summarized in Table III.
Conformity Assessment Routes for IVD Medical Devices

THE TRACEABILITY REQUIREMENT
The Directive requires that the values assigned to calibrators and/or control materials of IVD devices are traceable to available measurement standards and/or available measurement procedures of a higher order.
Annex I.A.3
The traceability of values assigned to calibrators and/or control materials must be assured through available reference measurement procedures and/or available reference materials of a higher order
How to achieve this traceability is the subject of the work in the technical committee CEN/TC 140 “In vitro diagnostic medical devices” of the European Committee for Standardization. In this committee a European Standard is developed in parallel with the technical committee ISO/TC 212 “Clinical laboratory testing and in vitro diagnostic test systems” of the International Standardization Organization. This European and International Standard is at present in the stage of the pre draft prEN ISO 17511.
The usual end point of a traceability chain is a national or international measurement standard, which ideally has a value traceable to the SI system of units. In order to make a measurement traceable to the SI, the measurable quantity must be clearly defined. For those measurements for which traceability to the SI is not (yet), possible, it is also necessary for the manufacturer to precisely (exactly), define the intended use of the product e.g., in relation to medical needs.
Traceability to the SI requires a complete knowledge of the molecular structure of the analyte, and a definition of the quantity and the unit. For example, the amount of substance concentration of cortisol in human serum, with a unit of mol.m3 or mol.l-1.
CGPM = Conference Generale des Poids et Mesures, NMI = National Metrology Institute, ARML = Accredited Reference Medical Laboratory, ML = Medical Laboratory.
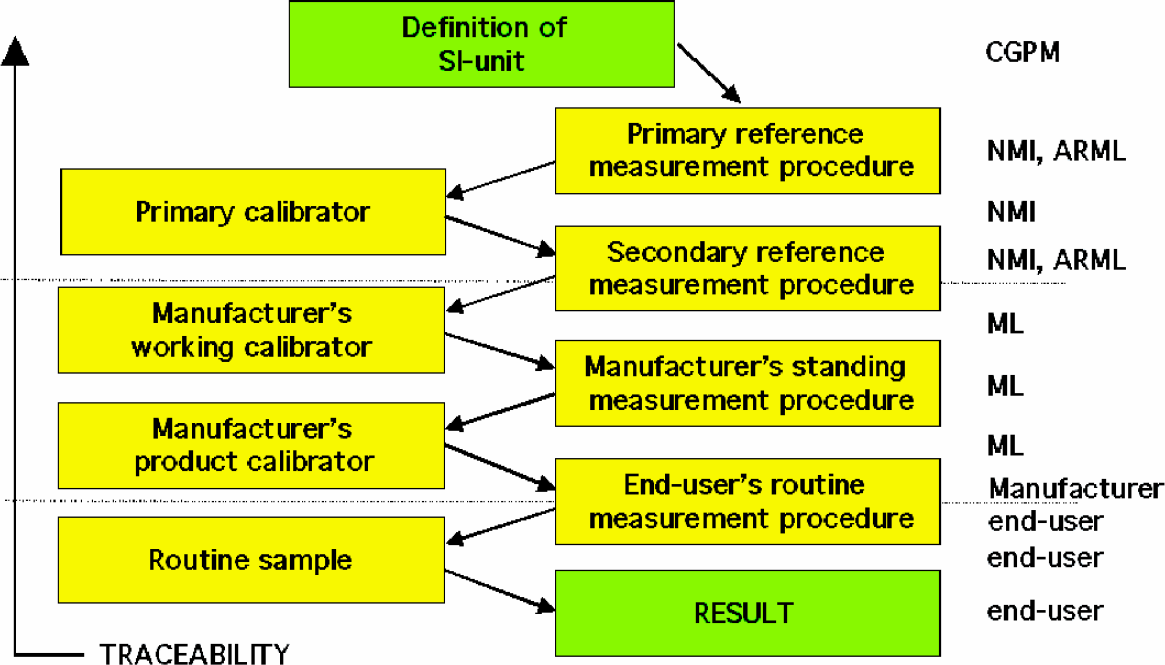
In order to assign values to measurement standards for quantities of which measurement results can be made traceable to SI, a primary measurement procedure must be available, which is a measurement method specific for the analyte and whose measurement result can be obtained without reference to a standard of the same kind. Isotope ratio mass spectrometry, coulometry, and gravimetric and volumetric analysis have the potential to be primary, depending on the specificity of the measurement procedure and quantitative knowledge of all influence factors. A reference material which is used as a calibrator and whose value is assigned by a primary method is called a primary reference material. Primary reference measurement procedures and one or more primary reference materials (used as calibrators) are available for approximately 25–30 types of well defined compounds such as some electrolytes, metabolites, steroid hormones, and some thyroid hormones.
A complete traceability chain for such well-defined analytes, being the inverse of the calibration hierarchy, starts with the measurement result as obtained with the IVD device up to the SI.
For other quantities the traceability chain will stop before the SI level and the measurement result will become traceable to, for example, an arbitrary unit. The prEN ISO 17511 distinguishes four cases for which traceability can be achieved up to:
An international conventional reference measurement procedure (RMP), and one or more international conventional calibration materials with values assigned by that procedure (e.g., Haemoglobin A1c).
An international conventional RMP but no international conventional calibration materials are available. This category comprises ∼30 types of quantity with components such as haemostatic factors.
One or more international conventional calibration materials, but no international conventional RMPs are available. This category comprises ∼250 types of quantity, including quantities for which WHO international standards are available, such as e.g., (glyco), protein hormones.
An in-house measurement procedure and/or a calibrator of the manufacturer. Neither international conventional RMPs or calibration materials are available. This category comprises ∼300 types of quantity such as tumor markers and antibodies.
CONCLUSION
In the future, it will be necessary to improve traceability for results of all quantities for which traceability to SI is not possible and for which RMPs and/or reference materials are missing. This can be achieved only, by working together on an international level between National Metrology Institutes (NIST, NMi, PTB, etc.), accredited reference laboratories for laboratory medicine (ARMLs), and international scientific organizations to determine priorities and to start development of new primary or international conventional RMPs and calibrators. □